Because R is an exemplary environment in which to create data-driven graphics, RCy makes it easy to create and display expressive and detailed molecular maps.
Here is a simple example, a modest addition to our standard
'test and demo' RCy graph, which you can create via a call to makeSimpleGraph () (see 'methods'). There is an
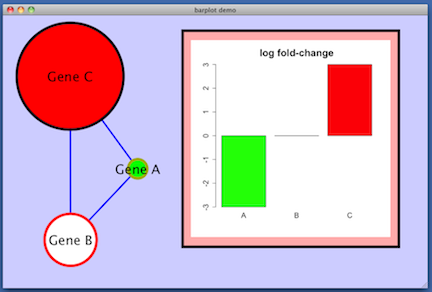
lfc attribute on the nodes, some made-up values (-3, 0, 3) which are used in this image to control the
color of the nodes. Red indicates positive lfc, white is zero lfc, and green is negative lfc.
For example: create a named list in R and display it as a barplot.
lfc = c (-3, 0, 3)
names (lfc) = c ('A', 'B', 'C')
barplot (lfc, main='log fold-change', cex.main=2, cex.axis=1.5, cex.names=1.5, col=c ('green', 'white', 'red'))
lfc = c (-3, 0, 3)
names (lfc) = c ('A', 'B', 'C')
png ('barplot.png')
barplot (lfc, main='log fold-change', cex.main=2, cex.axis=1.5, cex.names=1.5, col=c ('green', 'white', 'red'))
dev.off ()
window.title = 'barplot demo'
g = RCytoscape::makeSimpleGraph ()
g = graph::addNode ('lfc.plot', g) # this is the new informational node
nodeData (g, 'lfc.plot', attr='label') = 'plotting surface' # give it a good label, later hidden by the plot
cw = new.CytoscapeWindow (window.title, g)
hideAllPanels (cw)
displayGraph (cw)
layoutNetwork (cw, 'jgraph-spring')
setWindowSize (cw, 800, 600)
fitContent (cw)
setZoom (cw, 0.9 * getZoom (cw))
setNodeLabelRule (cw, 'label')
node.attribute.values = c ("kinase", "transcription factor")
colors = c ('#A0AA00', '#FF0000')
setDefaultNodeBorderWidth (cw, 5)
setNodeBorderColorRule (cw, 'type', node.attribute.values, colors, mode='lookup', default.color='#000000')
count.control.points = c (2, 30, 100)
sizes = c (20, 50, 100)
setNodeSizeRule (cw, 'count', count.control.points, sizes, mode='interpolate')
setNodeColorRule (cw, 'lfc', c (-3.0, 0.0, 3.0), c ('#00FF00', '#FFFFFF', '#FF0000'), mode='interpolate')
redraw (cw)
lfc.values = noa (g, 'lfc') [1:3] # don't pick up the fourth node -- that's the informational one!
png ('barplot.png')
barplot (lfc.values, main='log fold-change', cex.main=2, cex.axis=1.5, cex.names=1.5, col=c ('green', 'white', 'red'))
dev.off ()
# the image file is, for now, in your working directory, with the name you gave it
setNodeImageDirect (cw, 'lfc.plot', sprintf ('file://%s/%s', getwd (), 'barplot.png'))
setNodeSizeDirect (cw, 'lfc.plot', 200)
layoutNetwork (cw, 'force-directed')
redraw (cw)
setNodeImageDirect (cw, 'lfc.plot', 'file://data/proj/barplot.png') setNodeImageDirect (cw, 'lfc.plot', 'http://server.xyz.com/data/proj/barplot.png')